Introduction
At its core, ubiquity is a modeling language and a set of scripts
meant to facilitate model development and deployment. The focus of this
document is on the model description language. This is a plain text
file, referred to as a system file. Each line contains
a descriptor (e.g. <P>) which defines an aspect of
the model, and comments are made with the hash sign (#).
What follows is an overview of the different components of the language
that can be used to create a system file.
Parameters
System parameters <P>
Each system parameter is specified with a name, value, lower bound, upper bound, units, whether it should be editable in the ShinyApp and the ‘type’ of parameter (grouping in the ShinyApp). The values of eps (machine precision, smallest value that is not zero) and inf (infinity) can be used. For example to specify a parameter koffR with a value of .1 that is positive and a parameter KDR with a value of .04 that is also positive the following would be used:
# name value lb ub units editable type
<P> koffR 0.1 eps inf 1/hr yes Target
<P> KDR 0.04 eps inf nM yes TargetParameter sets
Often a model will be developed to incorporate different situations or scenarios. For example, a model may be used to describe both healthy and diseased individuals. When these differences are simply parametric in nature, it can be cumbersome to code a model multiple times (once for each parameterization). This framework provides a mechanism for including multiple parameterizations withing the same system file. Consider the system below where we want to describe antibody disposition. For humans this is described by a two compartment model, but for mice a single compartment is needed.
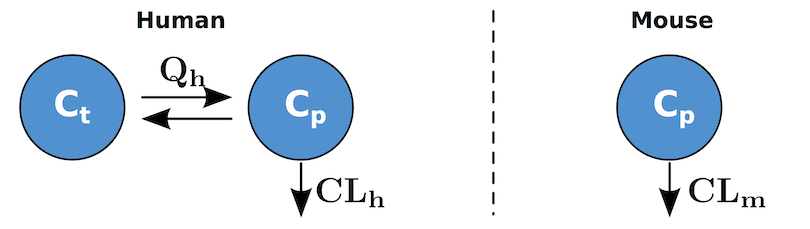
First we create a set of parameters describing the human scenario. These are the mean parameters taken from the literature [DM]:
<P> Weight 70.0 eps inf kg yes System # Organism weight
<P> CL 0.0129 eps inf L/hr yes System # Systemic Clearance
<P> Q 0.0329 eps inf L/hr yes System # Inter-compartmental clearance
<P> Vp 3.1 eps inf L yes System # Vol. central compartment
<P> Vt 2.8 eps inf L yes System # Vol. peripheral compartmentWhen a parameter is created using the <P>
descriptor it is part of the default parameter set. This is
the short name1 for a parameter set. A longer more verbose
name can be given as well, and this is what will be seen in the
ShinyApp. The human parameter set can be labeled using the
PSET descriptor in the following way:
<PSET:default> mAb in HumanWhere default is the parameter set short name name, and
“mAb in Human” is the value shown to the user in the ShinyApp.
Next, to add the parameterization for mice we simply create a new set in the following way:
<PSET:mouse> mAb in MouseThis alone would create a new parameter set with a short name
mouse, and is an exact copy of the default parameter set.
To identify the parametric differences between the mouse
and human we use PSET in the following
way:
<PSET:mouse:Weight > 0.020 # 20 gram mouse
<PSET:mouse:CL > 7.71e-6
<PSET:mouse:Q> 0.0
<PSET:mouse:Vp > 1.6e-3
<PSET:mouse:Vt > 1 # arbitraryConsider the clearance parameter entry where we want the murine half-life of an antibody [VR]:
<PSET:mouse:CL> 7.71e-6We use the set name (mouse) and the parameter name
(CL) and then we overwrite the default with the specified
value 7.71e-6. The other aspects of the parameter (bounds,
edit flag, etc.) will be the same as the default value.
Secondary parameters <As> and
<Ad>
A static secondary parameter refers to parameter does not change
during a simulation. These are specified using the
<As> descriptor and can be written in terms of system
parameters or previously defined static secondary parameters. These can
be used in differential equations, defining initial conditions, input
scaling and model outputs. This is similar to secondary parameters
defined in the $PK block in NONMEM. For example, if you
wanted to define the rate of elimination in terms of the system
parameters for clearance CL and volume of distribution
Vp the following would be used:
<As> kel = CL/VpA dynamic secondary parameters refers to a parameter that can change
during a simulation. This typically means it is defined, using the
<Ad> descriptor, in terms of a state or another
dynamic secondary parameter. These can be used in differential equations
and model outputs. These are similar to the parameters defined in the
$DES block in NONMEM. For example if you wanted to use the
concentration in the central compartment Cp but it was
dependent on the amount in that compartment Ap and the
volume of that compartment Vp the following would be
used:
<Ad> Cp = Ap/VpVariance parameters <VP>
Variance parameters are specified using the same format as system
parameters (<P>) :
# name value lower_bound upper_bound units editable grouping
<VP > SLOPE 0.01 eps inf 1/hr yes VarianceThe difference being that the <VP> descriptor is
used and that the grouping is set to Variance. These are
used when performing parameter estimation and when simulating with
residual variability.
Parameter estimation information <EST:?>?
Currently applies only to nlmixr2/rxode . Essentially all the parameters not listed here will be fixed.
This descriptor specifies information about parameters for
estimation. Sometimes it is necessary to estimate parameters in the log
space. Here you can specify the parmaeters to log transform (LT). If you
wanted to log transform parameters P1, P2, and
P3 you would do the following:
<EST:LT> P1; P2; P3NOTE That for the Monolix outputs to work correctly you will need to logtransform any parameters that have log-normal IIV assigned.
By default all parameters will be specified for estimation. If you
want to estimate a subset of parameters (P), say P1 and
P2, you can use the following:
<EST:P> P1; P2Variability: defining the variance/covariance Matrix
<IIV:?>? & <IIVCOR:?>?
Any variable name assigned to inter-individual variability (IIV) or
correlation/covariance (IIVCOR) term that makes sense to the user may be
used. The following sample codes have variable names (eg: ETACL) that
most likely make sense to a population modeler or NONMEM user. To define
an IIV term named ETACL with a variance of
0.15 use the following:
<IIV:ETACL> 0.15The next we need to associate this IIV term with a system parameter.
To associate this IIV term with the clearance (system parameter
CL) and specify that it has a log normal distribution
(LN) we would simply write:
<IIV:ETACL:LN> CLAlternatively a normal (N) distribution can be used.
Next we specify the IIV term ETAV with a variance of
0.1. This IIV term also has a log normal distribution and
is applied to the parameter V:
<IIV:ETAV> 0.10
<IIV:ETAV:LN> VNow we can define the covariance (off-diagonal elements) between
CL and V to be 0.01 by using:
<IIVCOR:ETAV:ETACL> 0.01The order isn’t important and the IIV terms can be reversed
IIV and parameter sets <IIVSET:?>? &
<IIVCORSET:?>?
By default all parameter sets will have inter individual variability
specified using the <IIV> and
<IIVCOR> descriptors. To associate a specific set of
IIVs to a parameter set use the <IIVSET> and
<IIVCORSET> descriptors. These set descriptors
operate differently than the parameter set descriptors
(<PSET>). The <PSET> just
overwrites the default values and inherits the default
variance/covariance information. If you alter the IIV information for a
parameter set it will reset the IIV information for that
parameter set. The entire variance covariance matrix will need to be
specified for that parameter set.
If the parameter set MYPSET has been defined then the
following could be used to define the IIV for the parameters
Q and CL:
<IIVSET:MYPSET:ETAQ> 0.05
<IIVSET:MYPSET:ETAQ:LN> Q
<IIVSET:MYPSET:ETACL> 0.25
<IIVSET:MYPSET:ETACL:LN> CL
<IIVCORSET:MYPSET:ETAQ:ETACL> 0.01All the other system parameters will have no IIV information for this parameter set.
Differential equations
The differential equations in the system can be defined by simply writing them out. Alternative they can ‘built’ by using the different descriptors provided below. Part of the flexibility of ubiquity lies in the ability to combine these different notations. To construct a model (see section below: Bringing it all together) any combination of the five following methods can be used:
- Differential equations
<ODE:?> - Reaction rates
=?=> - Equilibrium relationships
<=kforward:kreverse=> - Sources and sinks
<S:?> - Movement between compartments
<C>
Writing ODEs <ODE:?>
Portions of differential equations can be specified here where ? is
the state or compartment. To define dA/dT as
koffR*C - konR*A*B we would write:
<ODE:A> koffR*C - konR*A*B It might be more convenient to specify an ODE across several lines, making things more readable. Just use multiple statements and they will be appended together. This would give the same result as the example above:
<ODE:A> koffR*C
<ODE:A> - konR*A*B Rate equations =?=>
It may be more convenient to write out chemical reactions rather than differential equations. This can be done using the general form:
[CR1]Reactant1 + [CR2]Reactant2 + ... =kf=> [CP1]Product1 + [CP2]Product2 + ...Where the stoichiometric coefficients, beginning with CR
and CP above, in brackets only need to be specified if they
are not one. The reaction order will be assumed to be equal to the
stoichiometric coefficient of the reactant. For a more specific example
Consider decomposition of hydrogen peroxide into water and oxygen:
H_2O_2 \xrightarrow{k_{deg}} H_2O + \frac{1}{2}O_2
In the system format this would be written in the following manner:
H2O2 =kdeg=> H2O + [0.5]O2And this will be translated in to the following differential equations:
\frac{dH_2O_2}{dt}=-k_{deg}H_2O_2 \\ \frac{dH_2O}{dt}= k_{deg}H_2O_2 \\ \frac{dO_2}{dt}= 0.5k_{deg}H_2O_2
Which could also be defined as differential equations using the
<ODE:?>?. This is the equivalent:
<ODE:H2O2> - kdeg*H2O2
<ODE:H2O> kdeg*H2O2
<ODE:O2> 0.5*kdeg*H2O2 The rates (e.g. kdeg) need to be defined as either a
system or secondary parameter. This is where you can put saturable
terms, such as Michaelis-Menten kinetics.
Equilibrium relationships
<=kforward:kreverse=>
Forward and reverse reaction rates can be written separately:
A + B =konR=> C
C =koffR=> A + BOr these can be written as equilibrium equations with the forward
(konR) and reverse (koffR) rates specified
as:
A + B <=konR:koffR=> C To specify this reaction as differential equations, the following could have also been used:
<ODE:A> koffR*C - konR*A*B
<ODE:B> koffR*C - konR*A*B
<ODE:C> -koffR*C + konR*A*BThe stoichiometric coefficients also define the reaction order here. For example, to create the following equilibrium reaction:
2A + 3B \mathop{\rightleftarrows}^{\mathrm{k_f}}_{\mathrm{k_r}} 4C
This rate notation could be used in the system file:
[2]A + [3]B <=kf:kr=> [4]C Which will produce the following in terms of differential equations:
\frac{dA}{dt} = 2k_rC^4 - 2k_fA^2B^3 \\ \frac{dB}{dt} = 3k_rC^4 - 3k_fA^2B^3 \\ \frac{dC}{dt} =-4k_rC^4 + 4k_fA^2B^3 To write this equilibrium reaction as differential equations the following would be used:
<ODE:A> = 2*kr*SIMINT_POWER[C][4] - 2*kf*SIMINT_POWER[A][2]*SIMINT_POWER[B][3]
<ODE:B> = 3*kr*SIMINT_POWER[C][4] - 3*kf*SIMINT_POWER[A][2]*SIMINT_POWER[B][3]
<ODE:C> = -4*kr*SIMINT_POWER[C][4] + 4*kf*SIMINT_POWER[A][2]*SIMINT_POWER[B][3]See below about generic functions such as
SIMINT_POWER[][].
Sources and sinks <S:?>
This method allows turnover to be described in terms of synthesis and
degradation terms. If A is produced at a rate of
ksynA (mass quantities), degraded at a rate of
kdegA, and modeled in concentration units then the sources
are specified on the left hand side of <S:?> and the
sinks (elimination) are specified on the left hand side of
<S:?>. Multiple sources and sinks can be separated by
semicolons. In this example with a compartment volume V
ksynA/V <S:A> kdeg*AWhich is the same as writing out the differential equation:
<ODE:A> koffR*C - konR*A*B + ksynA/V - kdeg*AMovement between compartments <C>
When mass moves between two physical spaces with different volumes we
need to specify, for each compartment, the species, volume and rate of
transport. The <C> descriptor allows us to just
identify the compartment information separated by semicolons (order is
important)
Species; Volume; Rate <C> Species; Volume; RateFor movement between a central compartment A with a
volume V to the tissue space At with a volume
Vt at rates kps and ksp
respectively this is specified in the following manner:
A; V; kps <C> At; Vt; kspWhich is the equivalent of the following differential equation:
<ODE:A> -kps*A + ksp*At*Vt/V
<ODE:At> +kps*A*V/Vt - ksp*AtBringing it all together
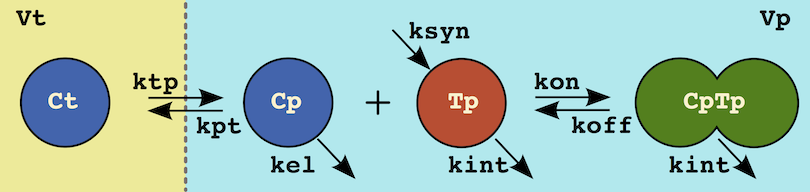
As a final example consider the target-mediated drug disposition system above. This system can be defined with a set of ODES:
<ODE:Ct> Cp*kpt*Vp/Vt - Ct*ktp
<ODE:Cp> -Cp*kpt + Ct*ktp*Vt/Vp - kel*Cp + koff*CpTp - kon*Cp*Tp
<ODE:Tp> + ksyn/Vp - kint*Tp + koff*CpTp - kon*Cp*Tp
<ODE:CpTp> - kint*CpTp - koff*CpTp + kon*Cp*TpOr it could simply be defined in terms of the underlying processes:
# tissue distribution
Ct; Vt; ktp <C> Cp; Vp; kpt
# equilibrium
Cp + Tp <=kon:koff=> CpTp
# Turnover
ksyn/Vp <S:Tp> kint*Tp
<S:Cp> kel*Cp
<S:CpTp> kint*CpTpInitial conditions <I>
By default all initial conditions are zero. You can specify a
non-zero initial condition using the <I> string to
set a ‘state’ to a ‘value’
<I> state = valueValue can be any combination of numbers, system parameters
<P> or static secondary parameters
<As. Consider a turnover system where the value of
ksyn and kdeg are specified as parameters:
<P> ksyn 0.1 eps inf 1/hr yes Target
<P> kdeg 0.04 eps inf nM yes TargetWe can calculate the initial value for a target as:
<As> T_IC = ksyn/kdegThen we can specify the initial value of the target as:
<I> T = T_ICModel inputs
Inputs into the model include typical interventions such as bolus dosing or continuous infusions. However inputs here refers to mathematical inputs. Typically covariates may be attributes of the system (such as gender, or a specific genotype), but are treated here as inputs. When defining inputs it is necessary to provide typical/placeholder values. These provide default values for both the ShinyApp interface as well as the scripting level (Matlab and R) where they can be overwritten by the user.
Bolus dosing <B:times>,
<B:events>
The <B:?> descriptor is used to define bolus
dosing. Dosing information is broken down into a list of
times when bolus injections occur and a list of
events containing the amount the specified compartment will
receive.
Each of these has a scale that is used to convert the
bolus dosing information from proscribed units (mg daily) into the units
in which the system is coded (mg/mL and hours). So if dosing is done on
days 0, 1, 2… but the simulation time is hours, then the scale for the
dosing times is 24.The events contain the magnitude of the bolus at a
given time. If you want to dose into a central compartment
Cp in mg/kg and your central compartment is mg/mL then you
need to scale by the body weight (e.g. 70 kg) and the volume of your
central compartment (system or static secondary parameter
Vp) then the scale is 70/Vp.
If you just want to create a palceholder you can do the following:
# type state values scale units
<B:times>; [0]; 24; days
<B:events>; Cp; [0]; 70/Vc; mpk If you want to setup default dosing for the shiny app or scripts, you can do somethign more complicated. If you have multiple compartments receiving a bolus, the times must include all times in which a bolus may be applied to the system. If a state does not receive a bolus on a particular time, its magnitude at that time is 0.
To illustrate this consider the following dosing schedule:
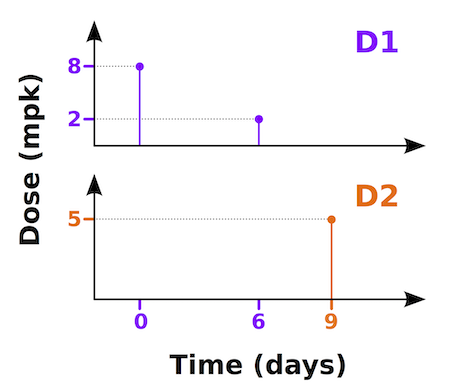
In this example we want to dose two different drugs into two
different states/compartments. Drug 1 (D1) will be dosed into
Cp1 and drug 2 (D2) into Cp2.
Dosing will be in mg/kg but concentrations are in mg/ml. The dosing time
is in days, but the simulation time units are hours. We will be dosing
D1 at 8 & 2 mpk on days 0 & 6. D2 will be dosed at 5 mpk on day 9.
# type state values scale units
<B:times>; [0 6 9]; 24; days
<B:events>; Cp1; [8 2 0]; 70/V1; mpk
<B:events>; Cp2; [0 0 5]; 70/V2; mpk Assume V1 and V2 are the compartmental
volumes for D1 and
D2 in ml, and the
subject body weight is 70 kg. This would convert those doses in mpk into
mg/ml. Again these are the default doses that can be overwritten
both in the ShinyApp and within scripts.
Continuous infusions <R:?>
Rates of infusion are defined using the <R:?>
descriptor. Like bolus values, infusion rates have two components. There
is a component that specifies switching times (e.g. switching from 10
mg/hr to 0). And each switching time has a corresponding rate of
infusion. This infusion rate will be held constant until the next time.
Also like the bolus specification there is a scale
associated with both infusion times and the
levels that converts the proscriptive units into the units
of the simulation. Consider the following example:
# name time/levels values scale units
<R:myrate>; times; [0 30]; 1/60; min
<R:myrate>; levels; [1 0]; 60; mg/minThese two entries create the infusion rate called
myrate. This can be used in any of your system
specifications (e.g., <ODE:Cp> myrate/Vp). The first
row specifies the times when the rate is changed (0 and
30 minutes). If the system is coded in terms of hours, then
the scale of 1/60 must be used. The levels indicate a rate
of 1 mg/min that is switched off at 30
minutes. This has to be converted to mg/hr using the scale of
60. You can add as many paired rate entries as you need to
describe as many infusion interventions as necessary.
Note: If you just want a placeholder you can just
set both of the values to [0].
Simple covariates
For simulation purposes covariates (normally found in a dataset) need
to be defined. Covariates can be either constant or change with time.
The times values must be the same scale as the system. The
following defines the value for the covariate RACE:
<CV:RACE>; times; [0]; hours
<CV:RACE>; values; [1]; ethnicityCovariates can also change with time. In this case consider the
subject weight (WGT). It begins at 70 and
measurements are made at several time points.
<CV:WGT>; times; [ 0 1680 3360 5040 10080]; hours
<CV:WGT>; values; [70 65 60 58 56]; kg Next we can alter how the simulations will interpret these values by
setting the type of covariate. By default the weight will be linearly
interpolated (type = linear), however we can hold the
weight constant until the next measurement is encountered (last value
carried forward) by declaring the type as step
<CVTYPE:WGT> stepNow if the model was parameterized for male and female subjects we can define two parameter sets (as described above) to account for this:
<PSET:default> Male
<PSET:female> Female And the values for the covariate can be changed for the set ‘female’:
<CVSET:female:WGT>; times; [ 0 1680 3360 10080]
<CVSET:female:WGT>; values; [60 55 52 50]Complicated covariates
Detailed time course profiles can be created as well. For example, to create a covariate profile that is zero from time 0-1 and at time 1 it jumps to 8 and decreases at a rate of 1 per unit time until time 5 where it stays at the value 4 until time 12. It might have a profile like the following:
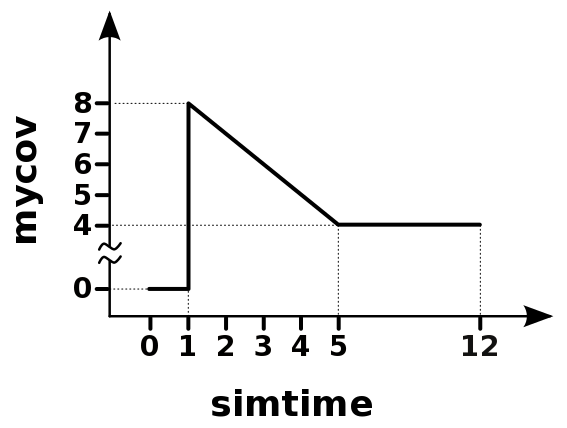
# name time/values values units
<CV:mycov> ; times; [0 .999 1 5 12]; hours
<CV:mycov> ; values; [0 0 8 4 4 ]; --
<CVTYPE:mycov> linearModel outputs
Outputs are defined here in terms of states, parameters, secondary parameters, input rates, and covariates listed above. The format used is:
<O> name = expressionFor example:
<O> A_obs = A
<O> Coverage = A/(KD + A)
<O> QC_CLtot = Cp*CL/Vp + Cp*Vmax/(Km+Cp)Outputs that begin with QC, like QC_CLtot
above, will not be displayed in the ShinyApp. This is intended to make
them available at the scripting level for quality control (QC)
purposes.
Output error model <OE:?> ?
This links parameters defined using <VP:?> to specific outputs
defined using
`<OE:OUTPUT> expression`The OUTPUT can be the name of any output defined with
<O>. The expression is a model type (add
for additive, and prop for proportional) with an equal sign and the name
of the variance paramter (<VP>) to use. To use more
than one error model type you separate the statments with ;
For example if you define the variance parameters add_err
and prop_err and want to use a proportional error model to
the output Cp you would use:
<OE:Cp> prop=prop_errTo use both additive and proportional error the following would work:
<OE:Cp> add=add_err; prop=prop_errThere are certain target-specific considerations. For example if you
are using Matlab or R and you just
wanted to do a weighted least squares estimation proportional to the
prediction squared you would use the following but fix
prop_err as 1.
<OE:Cp> prop=prop_errTo do least squares estimation you could do the following and fix
add_err as 1.
<OE:Cp> add=add_errThese represent the default values though it’s possible to overwrite them. In the Matlab and R workflows if you define the variance at the cohort level, the error models in the system file will be overwritten. ## Functions and operators
Most of the standard operators behave as expected (+,
-, *, & /) because most
languages use these consistently. There are however certain operators
and functions that differ between languages. For example, consider the
power function (a^b). In FORTRAN this
would be a**b, in Matlab it is a^b, and in C
it is pow(a,b). Given the objectives here (write once and
create multiple formats), this can be quite challenging. The solution
used here is to convert language specific functions and operators into
generic functions. So the power operator would be:
SIMINT_POWER[a][b]This would then be converted to the appropriate output format depending on the output target. The following generic functions can be used:
| Operator/Function | Example | Format |
|---|---|---|
| power | a^b | SIMINT_POWER[a][b] |
| exponential | e^a | SIMINT_EXP[a] |
| log base 10 | \log(a) | SIMINT_LOG10[a] |
| log base e | \ln(a) | SIMINT_LOGN[a] |
| less than | a < b | SIMINT_LT[a][b] |
| less than or equal | a \le b | SIMINT_LE[a][b] |
| greater than | a > b | SIMINT_GT[a][b] |
| greater than or equal | a \ge b | SIMINT_GE[a][b] |
| equal | a == b | SIMINT_EQ[a][b] |
| and | a \& b | SIMINT_AND[a][b] |
Current simulation time
For some systems you will want to access the simulation time. To do
this you can use the internal variable SIMINT_TIME.
Modeling quasi-equilibrium of target-mediated drugs
The free amount of a drug Cfree which is binding to a
receptor with an affinity of KD given total values
Ctot and Rtot the following can be calculated
using the following equation:
C_{free}=\frac{1}{2} \left[ (C_{tot}-R_{tot}-K_D) + \sqrt{(C_{tot}-R_{tot}-K_D)^2 + 4K_DC_{tot}} \right]
This can be tedious, especially using the generic functions above. To make this easier you can use the following generic function:
<Ad> Cfree = SIMINT_QEQ[Ctot][Rtot][KD]To get Rfree, just switch the first two arguments:
<Ad> Rfree = SIMINT_QEQ[Rtot][Ctot][KD]Timescales
Each system has default units in which it is constructed, and should be indicated in the comments of the model. It can be useful (for generating figures for example) to show simulations in different time scales. Now this can be achieved by multiplying the time outputs by the correct scaling factor. However this requires the end user to (1) remember the original timescale and (2) correctly scale that value.
Now while this is not particularly challenging from a mathematical
perspective, it introduces a chance for error. It is possible, instead,
to specify time scale information using the <TS:?>
descriptor. If the system is coded in hours, the following will define
timescales for the default (hours), days, weeks and months:
<TS:hours> 1.0
<TS:days> 1.0/24.0
<TS:weeks> 1.0/24.0/7.0
<TS:months> 1.0/24.0/7.0/4.0These are used both in the ShinyApp and at the command line in Matlab and R
Mathematical sets
Consider the following systems:
PBPK: Most of the organs in these systems are mathematically identical, with only variations in the parameters. However coding each of these organs or modifying an existing system (say to incorporate the presences of a target in each organ) can become tedious.
Anti-drug antibody generation: If we consider ADAs generated in response to therapeutic proteins, the response will consist of a distribution of ADAs in terms of their concentration and a separate distribution in terms of their affinity. Modeling this maturation process and the interactions between the ADAs, the therapeutic protein, and drug targets becomes unmanageable quickly.
The question is: How can we make difficult problems easy and intractable problems possible? The solution implemented here allows the system to be defined in terms of mathematical sets
Set notation <SET:?>?
Consider the interactions occurring in an assay designed to detect
drug (D) present in serum. In this assay a biotinylated
target (TB) is used to pull down the drug and a labeled
target (TL) is the signaling molecule used. The assay will
provide a signal when a complex containing both TB and
TL are present (TB:D:TL or
TL:D:TB). Samples can contain soluble target as well
(TS) which can interfere with the assay. To model this
assay, the following interactions should be considered:
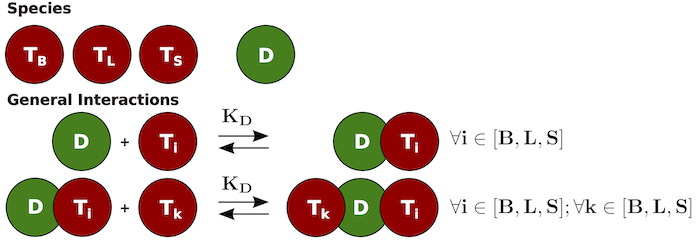
Several options are available to construct this system. The ODEs
could simply be typed out for every possible combination. It’s also
possible to use the equilibrium <=kon:koff=> for all
the interactions as well. However, there is another option that will
handle the enumeration more easily. First we define the two mathematical
sets TSi and TSk:
<SET:TSi> TL; TB; TS
<SET:TSk> TL; TB; TSWith these defined we can then use the curly brace notation
({ }) with any of the descriptors used to construct a
system. For example, the initial conditions for each of the target
states are defined as parameters (T0_TL,
T0_TS, T0_TB) in the model. These have to be
identified as initial conditions using the <I>
notation, and can be done with a single statement. This line:
<I> {TSi} = T0_{TSi} Expands to:
<I> TL = T0_TL
<I> TB = T0_TB
<I> TS = T0_TSSimilar to the initial condition, the equilibrium between the monomeric drug and the different targets can be defined using a single statement:
D + {TSi} <=kon:koff=> D_{TSi}That uses only one of the sets (TSi) and will be
expanded for each element in that set. To account for the formation of
complexes that contain a drug molecule and two different target
molecules, the following statement is used:
D_{TSi} + {TSk} <=kon:koff=> {TSk}_D_{TSi}This statement contains two different sets (TSi and
TSk). When multiple sets are encountered, every possible
combination is evaluated
Aligning Sets
By default sets will interpreted by evaluating every possible permutation as shown above. However, it may be desirable to pair or align sets. Take for example the transit compartments implemented by Lobo and Balthasar to delay the onset of drug effect on cancer cells [LB]. The transit compartment are implemented using a series of differential equations:
\frac{dK1}{dt} = (K-K1)\frac{1}{\tau} \\ \frac{dK2}{dt} = (K1-K2)\frac{1}{\tau} \\ \frac{dK3}{dt} = (K2-K3)\frac{1}{\tau} \\ \frac{dK4}{dt} = (K3-K4)\frac{1}{\tau}
It’s possible to code each of these individually, but it’s also possible to define these using mathematical set notation. We see that in the first equation K is paired or aligned with K1, and in the second it’s K1 and K2, etc. So first we define two sets of equal length whose elements are aligned:
<SET:TRIN> K; K1; K2; K3
<SET:TROUT> K1; K2; K3; K4 Next we write the <ODE:?>? statement in terms of
these sets, but we use the SIMINT_SET_ALIGN function:
SIMINT_SET_ALIGN[TRIN;TROUT][<ODE:{TROUT}> 1.0/tau*({TRIN}-{TROUT})]The first argument is the names of sets to align separated by a
semicolon ; and the second argument is the system file
descriptor written in terms of these sets. This will be expanded
internally into:
<ODE:K1> 1.0/tau*(K-K1)
<ODE:K2> 1.0/tau*(K1-K2)
<ODE:K3> 1.0/tau*(K2-K3)
<ODE:K4> 1.0/tau*(K3-K4)Set Functions
It can be useful to perform operations over sets. To do this you can use the following functions:
-
SIMINT_SET_SUMis the mathematical equivalent of \sum_{SET} -
SIMINT_SET_PRODUCTis the mathematical equivalent of \prod_{SET}
These functions take two bracketed arguments. The first argument is
the set name and the second argument is the mathematical relationship to
be expanded. For example, consider a system that has been parameterized
for several species. The variable EN_Mouse is 1 if the
species is mouse and 0 otherwise. Similarly for EN_Human,
EN_Monkey, etc. We have also defined the body weights as
system parameters: BW_Mouse for the mouse,
BW_Human for human, etc. Now we want to assign
BW to the value of the currently selected species (where
EN for that species is 1). First we would define the
species parameter set:
<SET:SP> Mouse; Rat; Monkey; HumanNext we would define the secondary parameter BW by
summing the product of the Boolean species parameter and the body weight
for that species:
<As> BW = SIMINT_SET_SUM[SP][EN_{SP}*BW_{SP}]Piecewise-continuous functions/parameters
<IF:?:?>
To specify a conditional assignment use the statement:
<IF:name:COND> boolean; valueHere name is the name of the secondary parameter be
defined and COND indicates that we have a Boolean condition
that needs to be satisfied. The condition (boolean) can be
and, or, greater than, etc. relationships. The parameter will be
assigned to have the value when this Boolean relationship is true. These
conditions can be a function of different elements of the system
depending on whether or not name refers to a static or dynamic secondary
parameter:
-
<As>function of system parameters, previously defined static secondary parameters and covariates that do not change for a given subject. -
<Ad>function of system parameters, static secondary parameters, states, previously defined dynamic secondary parameters and covariates.
It is important to include a default ELSE condition:
<IF:name:ELSE> valueConstructing a piece-wise continuous function/parameter
To see an example use the following command (use
?system_new to see a list of the available system file
examples):
system_new(system_file="pwc", file_name="system-pwc.txt")
In this example we specify fast (kelf) and slow
(kels) rates of elimination. We want to have the fast rate
be active when the drug concentration is above Cth and the
time is below Tf. The system parameters would look
like:
<P> kelf 1.0 eps inf 1/time yes System
<P> kels 0.01 eps inf 1/time yes System
<P> Cth 10 eps inf conc yes System
<P> Tf 10 eps inf time yes SystemNow we need to define the rate of elimination such that the
constraints above are followed. First we define kel as a
dynamic secondary parameter with a value of 0.0. Then we define the
different conditions and relevant values:
<Ad > kel = 0.0
<IF:kel:COND> SIMINT_AND[SIMINT_LT[SIMINT_TIME][Tf]][ SIMINT_GT[Cp][Cth]]; kelf
<IF:kel:ELSE> kelsControlling indices
By default, the build script will construct odes, parameter sets,
etc. automatically. This means that the order of states/compartments are
going to be arbitrary. Sometimes it is necessary to specify the order of
your states or outputs. For example when using NONMEM the order in the
control stream must correlate with the values in the dataset. To specify
that a state Cp should be compartment 3, the
following should be used:
<INDEX:STATE:Cp> 3The general notation for an output or state name
assigned to a number is:
<INDEX:STATE:name> number
<INDEX:OUTPUT:name> numberConcentrations vs amounts
It’s more convenient to model systems in terms of concentrations.
However some software does not allow scaling of inputs. And when inputs
are provided in mass units, you need your differential equation to also
be in mass units. You can use the <AMTIFY> descriptor
in your system.txt to tell ubiquity to generate perform
this conversion on the differential equations.
For example, if you defined the state Cc but want it to
be Ac within the nlmixr2 model target.
These are related by Cc = Ac/Vc and Vc is a
parameter the following would be used:
<AMTIFY> Cc; Ac; VcOptions
Several options can be specified using the <OPT:?
delimiter. If you’ve defined the days timescale using
<TS:?>? this can be used as the timescale for
plotting in the ShinyApp by using:
<OPT:TS> daysTo define the default output times for the ShinyApp and simulation scripts use the following:
<OPT:output_times> SIMINT_SEQ[0][100][1]Example system files
These example system files can be found in the examples directory of
the stand-alone analysis template (ubiquity_template.zip).
Most can also be loaded form the R package (see the help for
?system_new).
-
system-adapt.txt- Parent/metabolite structural model taken from the ADAPT5 Users Manual [ADAPT]. This system is used in the estimation examples of the ubiquity workshop. -
system-glp_study.txt- PK model parameterized for humans and NHPs, used as an example for GLP tox study design. -
system-in_vitro.txt- System to demonstrate in vitro/steady state modeling. See the estimation of static systems vignette for more details. -
system-mab_pk.txt- Linear model of mAb PK for humans taken from [DM]. This model is used in the simulation examples of the ubiquity workshop. -
system-one_cmt_cl.txt- One compartment model parameterized in terms of clearances and volumes. -
system-one_cmt_micro.txt- One compartment model parameterized for micro constants (rates). -
system-pbpk.txt- Implementation of large molecule PBPK model by Shah and Betts [SB]. This model provides good examples of how to use mathematical set notation. -
system-pbpk_template.txt- Template containing the species parameter from [SB]. This can be used to construct systems parameterized for multiple species. -
system-pwc.txt- Example of how to construct piece-wise secondary parameters (i.e. using if/then/else statements). -
system-sets.txt- Example of how to parameterized systems with multiple parameter sets. -
system-tmdd.txt- Example of how to code a TMDD model using either ODEs or process descriptors. -
system-tumor.txt- Implementation of transit effect model in cancer cell inhibition from [LB]. This demonstrates how to use aligned mathematical sets. -
system-two_cmt_cl.txt- Two compartment model parameterized in terms of clearances and volumes -
system-two_cmt_micro.txt- Two compartment model parameterized in terms of micro constants (rates)
Templates
When the system is built, multiple files are generated in the
temporary directory (transient) to support different
software. In the R package you can access these and other templates
programatically (see the help for ?system_fetch_template).
This is a list of the supporting software and what is generated.
R workflow
-
auto_simulation_driver.R: R-Script with named with placeholders used to run simulations. -
auto_analysis_estimation.R: R-Script with named with placeholders used to perform naive-pooled parameter estimation.
Matlab workflow
-
auto_simulation_driver.m: M-file with named with placeholders used to run simulations. -
auto_analysis_estimation.m: M-file with named with placeholders used to perform naive-pooled parameter estimation.
Other Software Targets
target_adapt_5.forandtarget_adapt_5-PSET.prm: The system defined for Adapt Fortran and a parameter (prm) file for each parameter setPSET.target_berkely_madonna-PSET.txt: This is a text file containing the system for the parameter setPSETto run in Berkeley Madonna.target_mrgsolve-PSET.cpp: This is a C++ file containing the system for the parameter setPSETto run using the inmrgsolvepackage in R.target_nlmixr-PSET.R: This R script defines the system for analysis innlmixr2for the parameter setPSET. Note: The nlmixr2 target can be used to create input model files for both NONMEM and Monolix
References
- [ADAPT] Adapt 5 Users Guide
- [DM] Dirks & Meibohm Clin. PK (2010) Oct 1;49(10):633-59
- [LB] Lobo, E.D. & Balthasar, J.P. AAPS J (2002) 4, 212-222
- [MK] Mager, D.E. & Krzyzanski, W. Pharm Res (2005) 22: 1589.
- [SB] Shah, D.K. & Betts, A.M. JPKPD (2012) 39 (1), 67-86
- [VR] Vieira & Rajewsky Eur J Immunol. (1988) Feb;18(2):313-6