Introduction
The workshop (workshop.ubiquity.tools) provides several examples of performing parameter estimation in ubiquity. To make a copy of these scripts and other supporting files in the current working directory run the following:
library(ubiquity)
fr = workshop_fetch(section="Estimation", overwrite=TRUE)-
analysis_parent.r- Least squares estimation of a single output -
analysis_parent_metabolite.r- Maximum likelihood estimation of two outputs -
analysis_parent_metabolite_global.r- Using global optimization packages
-
analysis_parent_metabolite_nm_data.r- Reading cohorts in from a NONMEM file
Each of these scripts build on the previous one to demonstrate
different features of the ubiquity parameter estimation routines. All of
these examples use a system (shown in the figure below) that contains
three differential equations tracking the mass of the parent drug in the
blood (Mpb) and in a tissue space (Mpt). In
the blood the parent can form (fmKp) a metabolite
(Mmb) with subsequent elimination (Km). The
parent/metabolite model was adapted from the ADAPT5 Users Manual (https://bmsr.usc.edu/files/2013/02/ADAPT5-User-Guide.pdf).

Using system_new (use ?system_new to see a
list of the available system file examples) you can copy this template
file into the current directory and build the system:
library(ubiquity)
system_new(file_name="system.txt", system_file="adapt", overwrite = TRUE)
cfg = build_system(system_file = "system.txt")Once the system has been built, you can create a local copy of an estimation template for the system:
system_fetch_template(cfg, template="Estimation")This should create the file: analysis_estimate.R in the
working directory. At the beginning of the script there are three
variables that are created to control what the script does and the
format of the output. The variable analysis_name defines
the prefix that will be prepended to the output generated by the script.
Archiving the analysis results is controlled by the Boolean variable
archive_results. Lastly, the script is controlled using the
flowctl variable (the possible options are listed and
commented out).
The estimation script has the following main components:
- Select the parameter set and subset of parameters to estimate
- Set options (simulation, estimation, etc)
- Load datasets
- Define cohorts
- Estimate parameters
- Plot results
These will be explored using the scripts above.
Least squares estimation/single output
(analysis_parent.r)
The first example above (analysis_parent.r) begins by
specifying that we want to perform a parameter estimation and archive
the results using the name1 parent_d1030 to indicate we
are analyzing the parent PK for the 10 and 30 mg dosing cohorts:
flowctl = 'estimate'
archive_results = TRUE
analysis_name = 'parent_d1030'Next we select the parameters to estimate. Because we’re only
estimating parent data, only the names of the parameters relevant to the
parent PK (pnames) are selected. To estimate all parameters
simply exclude the third argument. Because only system parameters are
being estimated a weighted least squares objective will be used2.
pnames = c('Vp', 'Vt', 'CLp', 'Q')
cfg = system_select_set(cfg, "default", pnames)Next we set options relevant to the estimation and the underlying simulation routines. In this example only the simulation output times have been specified, but any relevant parameters can be set. The template that is generated has several common options that are commented out. These can be uncommented and modified as needed.
cfg=system_set_option(cfg, group = "simulation",
option = "output_times",
seq(0,100,1))The dataset (shown in the table below) is included with the
ubiquity package and is accessed here using
system.file. In the example script it is referenced
explicitly. The format requirements of datasets is that they be flat
files with a header row. The format is flexible and only requires
time/observation information and columns required to filter the data and
isolate cohorts that have received the same treatment. The inputs are
defined when the cohorts themselves are defined. Data files are loaded
using system_load_data() and a name3 is assigned the
dataset. Data can be read from files (csv, xls, xlsx, and tab) or from
an existing data frame. This name (pm_data here) will be
used to reference this dataset later in the script.
cfg = system_load_data(cfg, dsname = "pm_data",
data_file = system.file("ubinc", "csv",
"pm_data.csv",
package = "ubiquity"))The dataset has a column for the observation time
(TIME), the subject id (ID), concentrations
for the parent (PT) and the metabolite (MT), a
BLQ flag and the nominal dose (DOSE). But only
some of these columns will be used (TIME, PT,
MT and DOSE), and any others
(BQL) will be ignored. This is intended to be an example
and not a general guide for a dataset format. It is not necessary to
have different outputs in different columns (wide format). The dataset
could be loaded in the tall skinny format as well. In this example we
are going to analyze the parent data for individuals given 10 or 30 mg
using a least squares objective.
Next we need to identify the data and inputs associated with those
data. This is done by defining cohorts, groups of data that
receive the same treatment and the model inputs (bolus dosing, infusion
rates, covariates) associated with those cohorts. In this example we
will define a cohort for each dosing group. To make sure we are starting
from a blank slate we can use system_clear_cohorts() to
remove any previously defined information.
cfg = system_clear_cohorts(cfg)For each cohort we define a list with information about that cohort.
Each cohort should have a unique name field4 and a
dataset field pointing to the dataset
(pm_data) loaded above. The optional cohort filter
(cf) field is used to reduce the entire dataset to the
records associated with this cohort. See the help for
system_define_cohort() for more information on how to
construct cohort filters.
cohort = list(
name = "dose_10",
cf = list(DOSE = c(10)),
inputs = NULL,
outputs = NULL,
dataset = "pm_data")Next it is necessary to define the inputs for this cohort. Here
inputs refers to model inputs which may include both dosing as well as
covariates, and the estimation template (generated using
system_fetch_template() above) should contain placeholders
for each of these defined in the system file. Note: For bolus and
infusion inputs, it is only necessary to define inputs that are nonzero,
and for covariates it is only necessary to define those that differ from
the definitions in the system file. For each input there is an
AMT field and TIME field and the units here
are those specified in the system file (both AMT and
TIME are internal indentifiers and not taken from the
dataset).
cohort[["inputs"]][["bolus"]] = list()
cohort[["inputs"]][["bolus"]][["Mpb"]] = list(TIME=NULL, AMT=NULL)
cohort[["inputs"]][["bolus"]][["Mpb"]][["TIME"]] = c( 0) # hours
cohort[["inputs"]][["bolus"]][["Mpb"]][["AMT"]] = c(10) # mpk Next we need to match the outputs in the model to the outputs in the
dataset. Under cohort$outputs there is a field used to
group each output. Here the cohort output mapping for
the blood PK of the parent output is Parent. The times and
observations in the dataset are found in the 'TIME’ column
and the ’PT’ column (missing data specified by -1 will be
dropped). These are mapped to the model timescale ('hours',
specified with <TS:?>) and model output
(’Cpblood’, specified with <O>). Note
the units in the dataset must be the same as those in the model:
cohort[["outputs"]][["Parent"]] = list()
# Mapping to data set
cohort[["outputs"]][["Parent"]][["obs"]] = list(
time = "TIME",
value = "PT",
missing = -1)
# Mapping to system file
cohort[["outputs"]][["Parent"]][["model"]] = list(
time = "hours",
value = "Cpblood",
variance = "1")
# Plot formatting
cohort[["outputs"]][["Parent"]][["options"]] = list(
marker_color = "black",
marker_shape = 1,
marker_line = 2 )For each output grouping in the cohort the marker color, shape and line type can be specified (controlling the plotted output).
cohort[["outputs"]][["Parent"]][["options"]] = list(
marker_color = "black",
marker_shape = 1,
marker_line = 2 )Finally the cohort is defined using
system_define_cohort():
cfg = system_define_cohort(cfg, cohort)We do the same thing for the 30 mg dose group:
cohort = list(
name = "dose_30",
cf = list(DOSE = c(30)),
dataset = "pm_data",
inputs = NULL,
outputs = NULL)
# Bolus inputs for the cohort
cohort[["inputs"]][["bolus"]] = list()
cohort[["inputs"]][["bolus"]][["Mpb"]] = list(TIME=NULL, AMT=NULL)
cohort[["inputs"]][["bolus"]][["Mpb"]][["TIME"]] = c( 0) # hours
cohort[["inputs"]][["bolus"]][["Mpb"]][["AMT"]] = c(30) # mpk
# Defining Parent output
cohort[["outputs"]][["Parent"]] = list()
# Mapping to data set
cohort[["outputs"]][["Parent"]][["obs"]] = list(
time = "TIME",
value = "PT",
missing = -1)
# Mapping to system file
cohort[["outputs"]][["Parent"]][["model"]] = list(
time = "hours",
value = "Cpblood",
variance = "1")
# Plot formatting
cohort[["outputs"]][["Parent"]][["options"]] = list(
marker_color = "red",
marker_shape = 2,
marker_line = 2 )
cfg = system_define_cohort(cfg, cohort)After the cohorts have been defined we call the estimation function
(system_estimate_parameters()). If flowctl is
set to 'plot previous estimate' or
'plot guess' the those values will just be returned.
pest = system_estimate_parameters(cfg,
flowctl = flowctl,
analysis_name = analysis_name,
archive_results = archive_results)If one of the estimation options are selected for the
flowctl then several files will be generated in the
output folder with the analysis_name as a
prefix:
-
output/parent_d1030-report.txt- Text file with a summary of the estimation results. -
output/parent_d1030-parameters_all.csv- Summary table with all parameters (estimated and fixed) -
output/parent_d1030-parameters_est.csv- Summary table with estimated parameters -
output/parent_d1030-system_update.txt- Text to update thesystem.txtfile with the new parameter estimates -
output/parent_d1030-sessionInfo.RData- The output ofsessionInfo()is stored in theSIobject in this data file
Next the system is simulated at the estimate and the data is stored
in erp.
cfg=system_set_option(cfg, group = "simulation",
option = "output_times",
seq(0,100,5))
erp = system_simulate_estimation_results(pest = pest, cfg = cfg) The information in this variable will be used to generate some
standard plots below, but it may be desirable to save this information
or generate your own figures. To do this it is necessary to understand
the structure of erp. This list has two different
fields.
-
erp$predData frame containing the time course data as well as the smooth predictions for all definedOUTPUTSfor a givenCOHORT. The columnSMOOTHis used to indicate what record type we’re dealing with. If theSMOOTHcolumn isFALSEthenOBScontains the observations andVARcontains the variance. IfSMOOTHisTRUEthenOBSandVARwill contain-1.-
TIME- Time in units of the data -
OBS- Observations (SMOOTH = FALSE), -1 (SMOOTH=TRUE) -
PRED- Predictions (SMOOTH = FALSE) -
VAR- Variance (SMOOTH = FALSE), -1 (SMOOTH=TRUE) -
SMOOTH-FALSEfor observation times,TRUEfor observations -
OUTPUT- name of the output -
COHORT- name of the cohort
-
-
erp$allList with model predictions for each output and state are generated for each cohort at the specified simulation times.-
ts.time- Simulation time scale -
ts.TS- An entry for each timescaleTS -
pred- Simulated predictions -
name- State or model output -
cohort- Cohort name
-
Lastly the predictions overlaying the data and the observed vs
predicted plots are generated using system_plot_cohorts().
Basic formatting of these figures is controlled using the
plot_opts list (see the ?system_plot_cohorts
for details).
plot_opts = c()
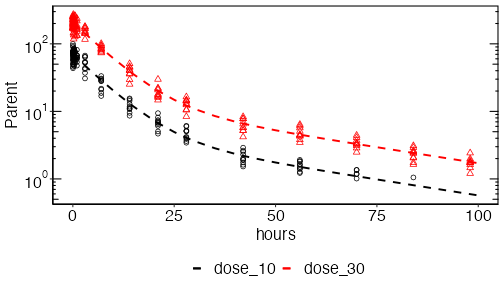
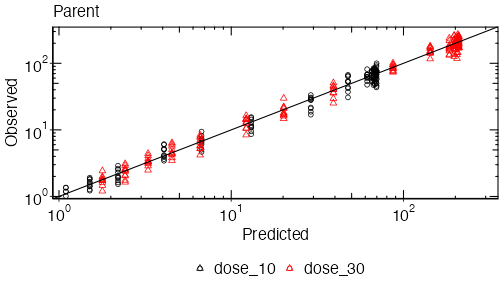
plot_opts$outputs$Parent$yscale = 'log'
plinfo = system_plot_cohorts(erp, plot_opts, cfg, analysis_name=analysis_name)When called, system_plot_cohorts() will write
png and pdf output for the time course and
observed vs predicted files. It will also return a list with the
ggplot objects and relative paths to the files as well. In
this example the following will be generated:
- output/parent_d1030_time course_Parent.pdf
- output/parent_d1030_time course_Parent.png
- output/parent_d1030_obs_pred_Parent.pdf
- output/parent_d1030_obs_pred_Parent.png
Automated Reporting
The outputs above provide components for generating presentations and
other documents. Coping and pasting these figures and tables into
documents can be tedious. It can be convenient to automate this process
and this is accomplished with the function
system_rpt_estimation().
PowerPoint
To append the results of an analysis to a PowerPoint document simply
initialize a new report (template="PowerPoint"), call
system_rpt_estimation() with the appropriate
analysis_name, and the results of the analysis will be
attached as slides.
cfg = system_rpt_read_template(cfg, template="PowerPoint")
cfg = system_rpt_estimation(cfg=cfg, analysis_name=analysis_name)
system_rpt_save_report(cfg=cfg,
output_file=file.path("output",paste(analysis_name, "-report.pptx", sep="")))This will then save the analysis results to the output
directory.
Word
The process for a Word document is the same. Just make sure that the
template is set to "Word" when the report is
initialized:
cfg = system_rpt_read_template(cfg, template="Word")
cfg = system_rpt_estimation(cfg=cfg, analysis_name=analysis_name)
system_rpt_save_report(cfg=cfg,
output_file=file.path("output",paste(analysis_name, "-report.docx", sep="")))For more information on integrated report generation see the
Reporting vignette.
Maximum likelihood/two outputs
(analysis_parent_metabolite.r)
This example is similar to the last except we are analyzing two different outputs (parent and metabolite) and we have a proportional variance model. So now we can estimate the parameters associated with those outputs as well as the variance parameters:
pnames = c('Vp',
'Vt',
'Vm',
'CLp',
'Q',
'CLm',
'slope_parent',
'slope_metabolite');
cfg = system_select_set(cfg, "default", pnames)The parameters being estimated contain variance parameters
(slope_parent and slope_metabolite) so a
maximum likelihood objective will be used. The cohort definitions look
much the same as those before except the variance model here is defined
as 'slope_parent*PRED^2', and there is a separate output
named Metabolite.
cohort = list(
name = "dose_10",
cf = list(DOSE = c(10)),
inputs = NULL,
outputs = NULL,
dataset = "pm_data")
# Bolus inputs for the cohort
cohort[["inputs"]][["bolus"]] = list()
cohort[["inputs"]][["bolus"]][["Mpb"]] = list(TIME=NULL, AMT=NULL)
cohort[["inputs"]][["bolus"]][["Mpb"]][["TIME"]] = c( 0) # hours
cohort[["inputs"]][["bolus"]][["Mpb"]][["AMT"]] = c(10) # mpk
# Defining Parent output
cohort[["outputs"]][["Parent"]] = list()
# Mapping to data set
cohort[["outputs"]][["Parent"]][["obs"]] = list(
time = "TIME",
value = "PT",
missing = -1)
# Mapping to system file
cohort[["outputs"]][["Parent"]][["model"]] = list(
time = "hours",
value = "Cpblood",
variance = "slope_parent*PRED^2")
# Plot formatting
cohort[["outputs"]][["Parent"]][["options"]] = list(
marker_color = "black",
marker_shape = 1,
marker_line = 1 )
# Defining Metabolite output
cohort[["outputs"]][["Metabolite"]] = list()
# Mapping to data set
cohort[["outputs"]][["Metabolite"]][["obs"]] = list(
time = "TIME",
value = "MT",
missing = -1)
# Mapping to system file
cohort[["outputs"]][["Metabolite"]][["model"]] = list(
time = "hours",
value = "Cmblood",
variance = "slope_metabolite*PRED^2")
# Plot formatting
cohort[["outputs"]][["Metabolite"]][["options"]] = list(
marker_color = "blue",
marker_shape = 1,
marker_line = 1 )
cfg = system_define_cohort(cfg, cohort)Similar modifications were made to the 30 mg dosing cohort.
Note: The variance model can be speified in the
system.txt file using the <OE:?>
descriptor. If you do not specify the variance in the cohort definition
the value from the system file will be used. If it is not specified in
either location then the default value (PRED^2) will be
used.
Global estimation routines
(analysis_parent_metabolite_global.r)
Now we build on the previous example to demonstrate how to select
different optimization routines. By default, the parameter estimation is
carried out using the Nelder-Mead optimization method from
the optim library. You can specify different functions in
the library. See the documentation for optim (?optim) for valid values
for method and elements for the control. For example, to use simulated
annealing change the method to SANN.
cfg = system_set_option(cfg, group = "estimation",
option = "method",
value = "SANN")There are also global optimization libraries in R, and several of these readily used with ubiquity. The settings for those are outlined below.
To use the particle swarm optimizer (pso package) set
the following options:
library(pso)
cfg = system_set_option(cfg, group = "estimation",
option = "optimizer",
value = "pso")
cfg = system_set_option(cfg, group = "estimation",
option = "method",
value = "psoptim")To use genetic algorithms (GA package) set the following
options:
library(GA)
cfg = system_set_option(cfg, group = "estimation",
option = "optimizer",
value = "ga")
cfg = system_set_option(cfg, group = "estimation",
option = "method",
value = "ga")
cfg = system_set_option(cfg, group = "estimation",
option = "control",
value = list(
maxiter = 10000,
optimArgs = list(method = "Nelder-Mead",
maxiter = 1000)))To use the constrained Markov Chain Monte Carlo (MCMC) from the
FME package set the following options:
library(FME)
cfg = system_set_option(cfg, group = "estimation",
option = "optimizer",
value = "fme")
cfg = system_set_option(cfg, group = "estimation",
option = "method",
value = "modMCMC")
# The control option here represents the arguments passed to the method above
# see the help for the method for valid options. Note that the following options
# are set internally and shouldn't be specified here:
# - f
# - p
# - lower
# - upper
cfg = system_set_option(cfg, group = "estimation",
option = "control",
value = list(
burninlength = 10,
niter = 500))The parallel partical swarm is implemented using the
ppso package. This is only available on github, so you’ll
have to install it from the repository there: https://github.com/TillF/ppso.
This package requiers the Rmpi package for parallelization.
library(ppso)
cfg = system_set_option(cfg, group = "estimation",
option = "optimizer",
value = "ppso")
# These are the methods in the pppso package:
# optim_pso, optim_pso_robust, optim_dds, optim_pdds_robust
cfg = system_set_option(cfg, group = "estimation",
option = "method",
value = "optim_pso")
# The control option here represents the arguments passed to the method above
# see the help for the method for valid options. Note that the following options
# are set internally and shouldn't be specified here:
# - objective_function
# - number_of_parameters
# - parameter_bounds
# - initial_estimates
# Need to install Rmpi
# https://fisher.stats.uwo.ca/faculty/yu/Rmpi/
cfg = system_set_option(cfg, group = "estimation",
option = "control",
value = list(
max_number_function_calls = 500,
max_number_of_iterations = 500,
number_of_particles = 5))The an alternative parallel partical swarm is implemented using the
parallelPSO package. This is only available on github, so
you’ll have to install it from the repository there. Use This patched
version: https://github.com/john-harrold/parallelPSO.
The original source is here: https://github.com/TillF/ppso.
This package requiers the doParallel package.
library(parallelPSO)
library(doParallel)
cfg = system_set_option(cfg, group = "estimation",
option = "optimizer",
value = "parallelpso")
cfg = system_set_option(cfg, group = "estimation",
option = "method",
value = "pso")
# The control option here represents the arguments passed to the method above
# see the help for the method for valid options. Note that the following options
# are set internally and shouldn't be specified here:
# - fitness_function
# - number_parameters
# - parameters_bounds
cfg = system_set_option(cfg, group = "estimation",
option = "control",
value = list(
max_number_iterations = 100,
number_of_partiples = 40,
parallel = TRUE))
cfg=system_set_option(cfg, group = "simulation",
option = "compute_cores",
value = detectCores() - 1)Note: Optimizers like SANN and the
global optimizers (pso, GA, etc.) are good for
identifying parameter sets outside of the region of the initial guess.
However, one consequence of these algorithms is they can quickly
approach the bounds. Consequently it is important to provide realistic
upper and lower bounds on the parameters (the <P>
descriptor in the system file or using system_set_guess()
at the scripting level). If you use the default value of machine
precision (eps) for the lower bound and infinity
(Inf) for the upper bound these optimization routines can
choose parameter values that can cause the internal simulations to
fail.
Cohorts from NONMEM dataset
(analysis_parent_metabolite_nm_data.r)
In the examples above, cohorts are defined manually. Sometimes you
may have data in a NONMEM dataset where the dosing information is
located in the dataset. It may be convenient to simply define a cohort
for each subject in the dataset. To do that the function
system_define_cohorts_nm can be used. The differences
between the script analysis_parent_metabolite.r will now be
highlighted:
First we load the NONMEM dataset and clear the cohorts:
cfg = system_load_data(cfg, dsname = "nm_pm_data",
data_file = system.file("ubinc", "csv",
"nm_data.csv",
package = "ubiquity"))
cfg = system_clear_cohorts(cfg);Next we define a filter to use on the dataset (include only the 10 and 30 mg doses):
For more information on filtering datasets see the help for
nm_select_records().
Now we define maps for the different outputs. For each output we
specify the variance, compartment (CMT) number, model
output and the missing number flag:
OBSMAP = list()
OBSMAP$PT = list(variance = 'slope_parent*PRED^2',
CMT = 1,
output = 'Cpblood',
missing = -1 )
OBSMAP$MT = list(variance = 'slope_metabolite*PRED^2',
CMT = 2,
output = 'Cmblood',
missing = -1 )Lastly we define a map for the model inputs. In this case we only
have a bolus in the Mpb compartment:
INPUTMAP = list()
INPUTMAP$bolus$Mpb$CMT_NUM = 1Unused columns in the dataset will be ignored. With the filter, input and observation maps defined, we now add the cohorts
cfg = system_define_cohorts_nm(cfg,
DS = 'nm_pm_data',
col_ID = 'ID', col_CMT = 'CMT', col_DV = 'DV',
col_TIME = 'TIME', col_AMT = 'AMT', col_RATE = 'RATE',
col_EVID = 'EVID', col_GROUP= 'DOSE',
filter = filter,
INPUTS = INPUTMAP,
OBS = OBSMAP,
group = FALSE)Contents of system.txt
#
# Parent/Metabolite example taken from Section 9.3 of the ADAPT5 Users Manual
#
# https://bmsr.usc.edu/files/2013/02/ADAPT5-User-Guide.pdf
#
<P> Vp 10.0 1e-5 100 L yes System
<P> Vt 10.0 1e-5 100 L yes System
<P> Vm 30.0 1e-5 100 L yes System
<P> CLp 1.0 1e-5 100 L/hr yes System
<P> CLm 1.0 1e-5 100 L/hr yes System
<P> Q 0.3 1e-5 100 L/hr yes System
<PSET:default> Original Estimates
<VP> slope_parent 0.1 1e-9 10 -- no Variance
<VP> slope_metabolite 0.1 1e-9 10 -- no Variance
<B:times>; [ 0 ]; 1; hours
<B:events>; Mpb; [ 0 ]; 70; mpk
<ODE:Mpb> -(CLp/Vp + Q/Vp)*Mpb + Q/Vt*Mpt
<ODE:Mpt> Q/Vp*Mpb - Q/Vt*Mpt
<ODE:Mmb> CLp/Vp*Mpb - CLm/Vm*Mmb
<O> Cpblood = Mpb/Vp
<O> Cmblood = Mmb/Vm
<TS:hours> 1.0
<TS:days> 1.0/24.0
<IIV:ETAVp> 0.08
<IIV:ETAVp:LN> Vp
<IIV:ETAVt> 0.08
<IIV:ETAVt:LN> Vt
<IIV:ETACLp> 0.08
<IIV:ETACLp:LN> CLp
<IIV:ETACLm> 0.08
<IIV:ETACLm:LN> CLm
<OPT:output_times> SIMINT_SEQ[0][100][1]
<DATA:HEADER:AUTOMATIC>